In a bioRxiv preprint, Chen et al. (2021) report the results of the first genomic sequencing of the virus.
Chen traces CCP back to RaTG13 and RmYN02 – strains reported by Chinese researchers as detected in Yunnan province of China, but yet to be found elsewhere.
The study considered over 4,000 full-length viral sequences made available by the Global Initiative for Sharing All Influenza Data (GISAID) EpiFlu database. Another 11 came from a Chinese database. Lastly, Chen analyzed ~2,61,000 genomes collected globally since the pandemic began. This comprises all the genomes in the database.
The researchers were able to identify distinct genotypes based on how commonplace certain mutations were. This helped trace superspreaders since they shaped the pandemic to a large extent. These individuals passed on specific genotypes with certain highly prevalent mutations. A single introduction of such genotypes led to an outbreak of infection, increasing evolution with spread.
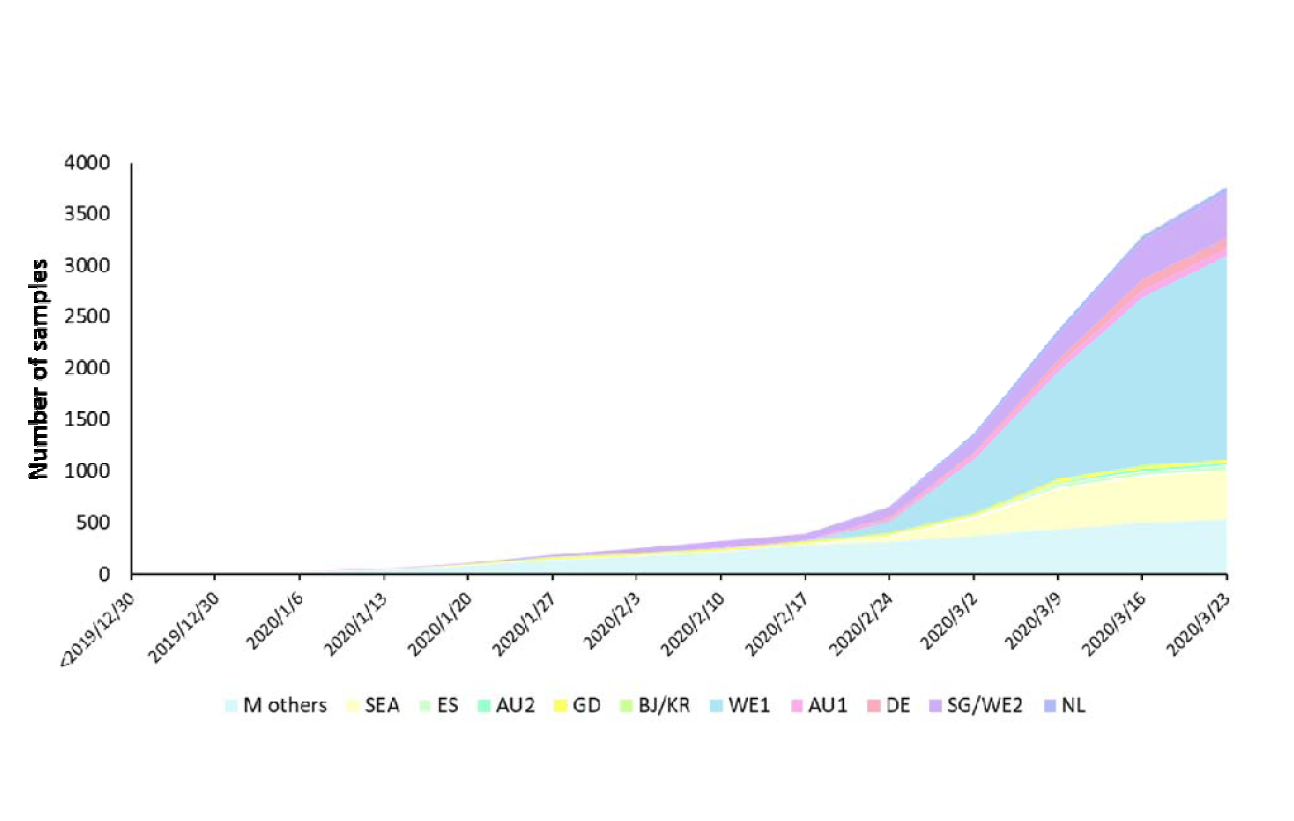
Six genotypes appear to have descended from the original strain with a single sequence – the M type variant originating in Wuhan – and responsible for more than 80% of the sequences in the study. Chen considers this as a true founder, and evident in spread to other regions of China before the Wuhan lockdown.
The six descendant genotypes are directly derived from the ancestral strain by characteristic mutations. The most prevalent genotype among these was the WE1 type, defined by four mutations. Three of the four defining mutations of the WE1 strain were found in three early samples collected in January 2020. Among WE1 genomes, 70% came from Western Europe (the UK, Iceland, Belgium, France, and the Netherlands, perhaps by traffic across the borders. It also made up ~35% of cases in the US.
The SEA type is the most common in the USA, however, but was isolated from three other countries, namely, Australia, Canada, and Iceland, indicating that cases from the USA had been imported there. This is also called the Washington State outbreak clade. The other four descendant genotypes were confined regionally.
The 34 sequences from early Wuhan cases showed two clusters, 30 belonging to the M type, but with extensive diversity. The remaining four formed another co-circulating cluster. Thus, at this early stage, there were 18 different genotypes among the 34 sequences.
In the USA, the prevalent strains belonged to the non-M types, probably from 12 cases imported from the Hubei province. These, in fact, were the earliest cases reported in the USA, with each showing a distinct genotype.
Half the US cases were SEA type, while ~35% were WE1. It indicates that the USA “endured the first wave of case importation from China and the second wave from Europe, which is consistent with the recent COVID-19 study of Washington State.” Among the 32 patients on the two cruise ships, the Grand Princess and the Diamond Princess, there were 25 different genotypes. This indicates that the virus mutates rapidly and extensively during person-to-person transmission.
The researchers developed a Strain of Origin (SOO) algorithm to match each genotype to its genome by mutational profile. When compared with mutation clustering, this approach showed a 90% agreement.
Using the same approach, they found that three of the top four GISAID clades were descendants of WE1. They estimated that one of three nucleotides in the viral RNA had undergone mutation over the 12 months of the pandemic.
They analyzed the top 100 mutations and generated a lineage-based pedigree chart. This story begins with a putative first case, supposed to be a patient with an ancestral SARS-CoV-2 genotype, and postulated to be present on November 17, 2019. This led to more infections. By January 1, 2020, the Huanan market was locked down, and 19 M type genome samples were documented.
However, the M type had already been incubating in the market for weeks, which accounts for the vast majority of genomes belonging to the M type at this time. With the expansion of the outbreak into Wuhan city at large, the city was locked down on January 23, 2020, with 80% of the viral genomes being of the M type. However, the Spring Festival had already prompted extensive travel to and from Wuhan, leading to the Chinese and then the global outbreak of COVID-19.
By April 7, 2020, more than 80% of cases worldwide were M type, but in September, 70% belonged to WE1, in three clades, namely, GR, G, and GH. The rise in M type continued, making up ~98% of cases by December 25, with almost 90% being caused by WE1 strains.
The researchers conclude that beginning with a single superspreader incident, the M type exploded over the world, following a few initial weeks when it passed unrecognized and uncontrolled. The M type acquired two concurrent mutations first, with another four defining mutations that led to the emergence of WE1 strains, and finally, another three that led to the WE1.1 strain. The rate of viral evolution, at ~27 substitutions per year, is not unusual, but the mechanism is still unclear.
Of the two new mutant strains attracting much attention, namely, the D614G point mutation and the N501Y mutation in the receptor-binding domain, both in the spike protein, are thought to be highly transmissible compared to the ancestral strain. The former was first documented in Western Europe in February 2020 and now makes up ~90% of strains, while the latter was first found in New York City on April 21, 2020, and makes up only 0.02% of cases.
Reference:
Chen, Y., Li, S., Wu, W., Geng, S., & Mao, M. (2021). Distinct mutations and lineages of SARS-CoV-2 virus in the early phase of COVID-19 pandemic and subsequent global expansion. doi:10.1101/2021.01.05.425339
Large-scale genome sequencing shows how SARS-CoV-2 mutated. (2021, January 10). Retrieved February 22, 2021, from News-medical.net website: https://www.news-medical.net/news/20210110/Large-scale-genome-sequencing-shows-how-SARS-CoV-2-mutated.aspx
