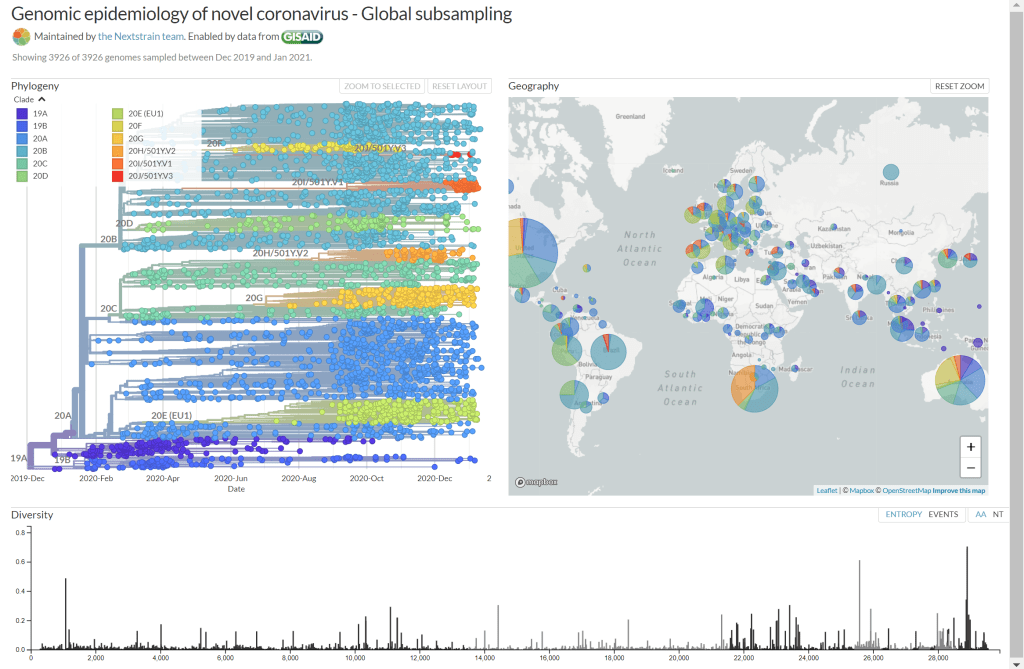
CoVid continues to be a remarkably stable genome.
Stability is consistent with an anthropogenic intervention and not consistent with a species jump.
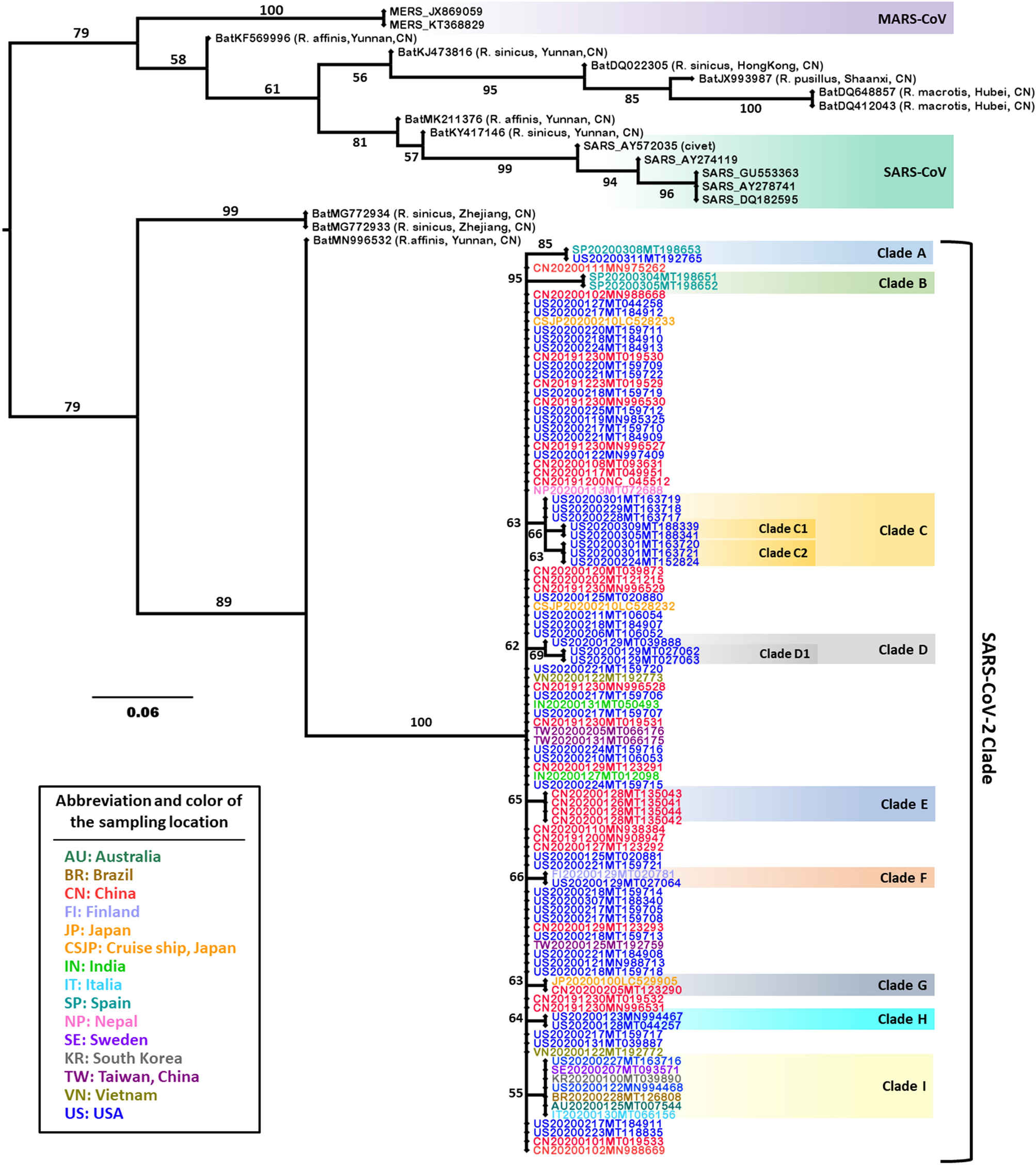
As reported in Nature (Li et al., 2020), MRP pseudo-sequence supertree analysis firmly disputes bat coronavirus RaTG13 as the last common ancestor of SARS-CoV-2, which was implied by other phylogenetic tree analysis based on viral genome sequences.
Li et al:
As the phylogenetic MRP pseudo-sequence supertree and ML tree exhibited, RaTG13 (MN996532), bat-SL-CoVZC45 (MG772933), bat-SL-CoVZXC21 (MG772934) and SARS-CoV-2s formed one major clade (Fig. 2, Supplementary Fig. S1). In particular, RaTG13 isolated from bat Rhinolophus affinis (Yunnan, China), is the closest relative of SARS-CoV-2s, which substantiates the previously reported phylogeny of SARS-CoV-2s constructed with the whole genome39,40. However, the phylogenetic distance of SARS-CoV-2s and RaTG13 was distinctly exhibited in the MRP pseudo-sequence supertree (Fig. 2); by contrast, it was barely observed in the phylogenetic ML tree constructed in this study (Supplementary Fig. S1) or previous report3
…
Whatsoever, the above distinct phylogenetic analysis results showed beyond a reasonable doubt that the rates of evolution on sequences of varied proteins in SARS-CoV-2s are highly non-uniform. There probably exists another bat coronavirus in divergent species as the adjacent ancestor of SARS-CoV-2, and/or SARS-CoV-2s already made advanced evolution in its animal host. Anyway, what is clear is that the actual validity of RaTG13 be the direct ancestor of SARS-CoV-2 is seriously questioned, although they share 96.5% identical genome sequence. Taking RaTG13 as the last common ancestor of SARS-CoV-2 would seriously mislead phylogenetic inference of SARS-CoV-2.
Also notable this week is the report in News-medical.net summarizing a recent bioRxiv* preprint study in which University of Hawaii researchers showed that the S gene of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is continuously mutating. Further, the researchers report phylogenetic analysis suggesting a unique strain with an emerging mutation in an altered spike glycoprotein may be present in Hawaii.
References:
Li, T., Liu, D., Yang, Y., Guo, J., Feng, Y., Zhang, X., … Feng, J. (2020). Phylogenetic supertree reveals detailed evolution of SARS-CoV-2. Scientific Reports, 10(1), 22366.
Study shows P681H mutation is becoming globally prevalent among SARS-CoV-2 sequences. (2021, January 10). Retrieved January 16, 2021, from News-medical.net website: https://www.news-medical.net/news/20210110/Study-shows-P681H-mutation-is-becoming-globally-prevalent-among-SARS-CoV-2-sequences.aspx
auspice. (n.d.). Retrieved January 16, 2021, from Nextstrain.org website: https://nextstrain.org/ncov/global
